Multiscale Modeling and Simulations of Chemical Reactions in Complex Environments
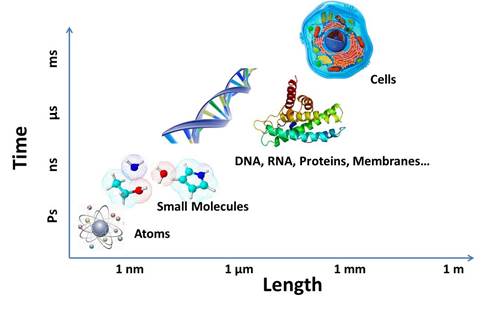
Many chemical and biological processes situate in complex environments, occur at various spatial and time scales, and require different levels of theory for proper descriptions.
Cutting-edge multiscale modeling and simulations help us explore molecular structures, reaction mechanisms, and underlying dynamics in the chemistry world. Many chemical reactions occur in complex environments, where the interactions and movements at different spatial and time scales often show distinct characteristics and require different levels of theory for proper treatments.
QM/MM methods describe a small primary region where reaction is taking place (e.g. an enzyme active site) by the sophisticated quantum theory and the interacting surroundings (e.g., protein matrix and bulk solvent) by classical molecular mechanics (Newtonian theory). By integrating the accurate quantum mechanics and the computationally efficient molecular mechanics, QM/MM offers realistic and effective descriptions for reactions in complex environments. (A textbook for beginners can be found here.)
Unfortunately, conventional QM/MM methods have two notable limitations. First, no partial charge transfer is allowed between the quantum and classical subsystems, even though such electron delocalization is likely (e.g. when the two subsystems are connected through hydrogen bonds or polar covalent bonds). Second, all atoms are designated into either subsystem before a simulation starts, and the partition will not change during the simulation. However, as the reaction proceeds, it might be necessary to dynamically reclassify the atoms according to their changing roles.
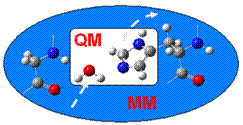
Our ultimate goal is an open boundary between the quantum and classical regions that allows on-the-fly exchange of partial charges and atoms in dynamics simulations.
We develop novel algorithms that overcome those critical limitations in conventional QM/MM. For example, we proposed the adaptive-partitioning QM/MM schemes that allow smooth transitions when atoms moving into or out of the quantum subsystem during molecular dynamics simulations. We also pioneered the flexible-boundary treatments that describe partial electron transfer between the quantum and classical subsystems based on chemical-potential equalization. The algorithms have been implemented in the computer software package QMMM.
Recently, in collaboration with Prof. Emilie Guidez, we extended the two-layer QM/MM model to multi-layer Q1/Q2/MM model (Q1 and Q2 are two distinct QM levels of theory). We proposed a "crazy" idea called "alchemical QM", where the basis sets, Fock matrices, and overlap matrices are interpolated between Q1 and Q2.
Our ultimate goal is to establish an open boundary that permits on-the-fly exchange of both partial charges and atoms between the quantum and classical regions. The open boundary effectively turns the finite-size quantum subsystem infinite and can in principle continuously describe behavior of interest through any length of time, thus holding the promise for the utilization of small quantum subsystems and high-levels of quantum theory, which can potentially lead to new insights.
We have applied quantum and classical calculations to study chemical and biological processes of interest.
One application is the solvation and transport of ions and reactive species in solvent and across artificial membranes. These processes are essential to the functioning of battery and to catalysis, which are important in the development of new energy resources and new materials.
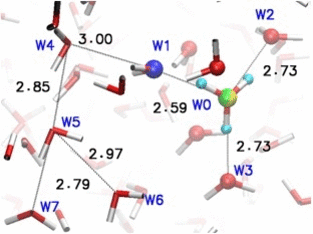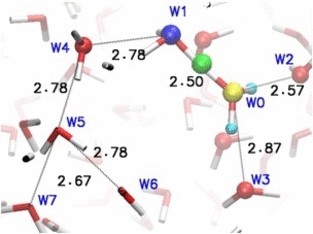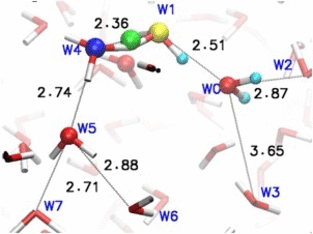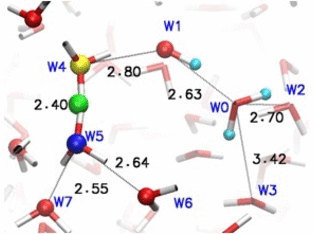
Adaptive quantum and classical dynamics simulations detail proton relay between water molecules in bulk solution. More recent development "adpative-shape" QM/MM keeps the QM zone as small as possible while as large as necessary.
Another application is ion pump and channel proteins. Currently, we are modeling translocations of ions through membrane by CLC chloride/proton, CLCF fluoride/proton, and NarK nitrate/nitrite transport proteins, which are associated with a variety of critical physiological and cellular processes such as neuroexcitation, cell-volume regulation, organic solute transport, and muscle contraction.
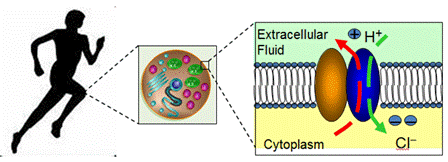
Regulating chloride ion and proton through membrane, CLC chloride ion transport proteins are critical for a variety of physiological and cellular processes.
In collaboration with experimental biochemist Prof. Jefferson Knight from our department, we also investigate how calcium ions trigger the docking and insertion of the C2A domain of synaptotagmin-7 into a membrane; the synaptotagmin-7 protein regulates insulin secretion in pancreatic beta cells and is an important target for drug design in diabetes therapy.
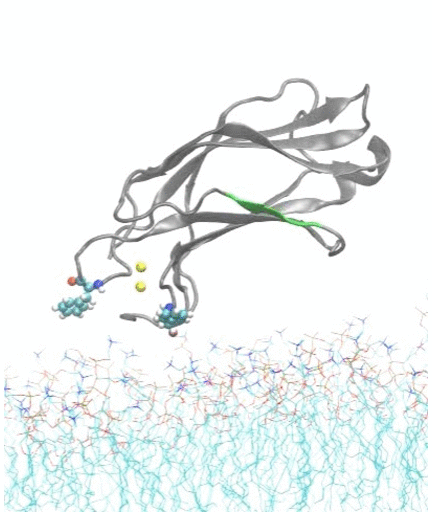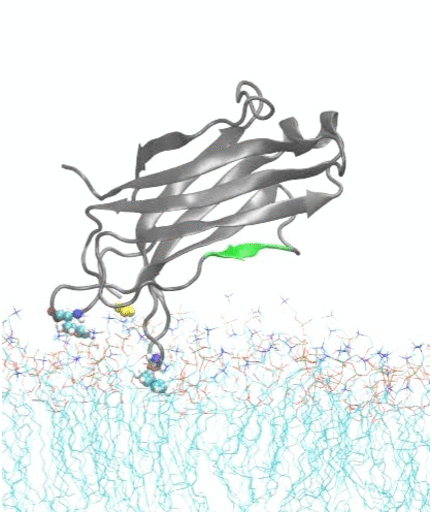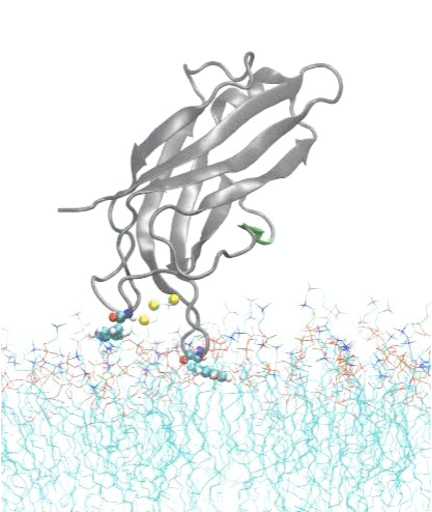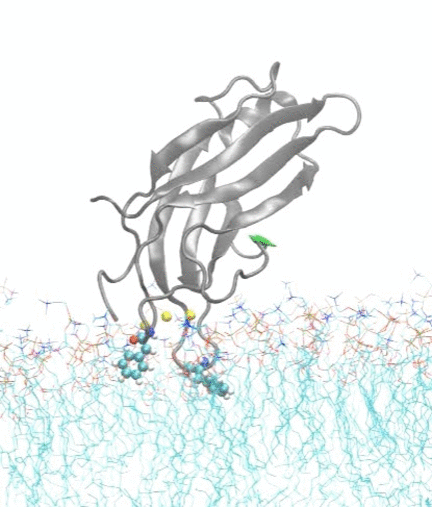
Molecular dynamics simulations show spontaneous insertion of the C2A domain of synaptotagmin-7 protein into a membrane.
Dr. Hai Lin

Professor, Chemistry Department,
University of Colorado Denver,
SI 4126B, Campus Mail 194,
PO Box 173364, Denver, CO 80217
Phone: +1-303-315-7656
Fax: +1-303-556-4776
E-mail
ORCID • Google Scholar • Researcher ID
Interested students are very welcomed to contact me via my email about research projects and research positions.
By the way, I found this interesting link.