Development of multilayer adaptive partitioning methods to model ion transport across protein channels.
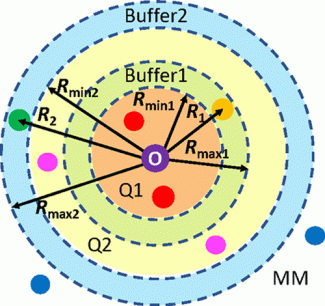 In collaboration with Pr. Hai Lin at CU Denver, our group aims to develop new multilayer adaptive partitioning methods in order to model molecule and ion transport in various systems such as the NarK protein. In these new methods, the substrate of interest and its immediate surroundings are modeled with high level electronic structure methods (Q1 zone). Residues that are further away are modeled with lowe level electronic structure methods (Q2 zone) and the rest of the system is modeled with molecular mechanics (MM). As the substrate moves, the atoms in the system are reclassified into the appropriate zone on the fly during molecular dynamics simulations. Buffer zones between adjacent layers allow a smooth extrapolation of energies and forces. This project combines method development, software implementation (GAMESS and QMMM) and application simulations.
In collaboration with Pr. Hai Lin at CU Denver, our group aims to develop new multilayer adaptive partitioning methods in order to model molecule and ion transport in various systems such as the NarK protein. In these new methods, the substrate of interest and its immediate surroundings are modeled with high level electronic structure methods (Q1 zone). Residues that are further away are modeled with lowe level electronic structure methods (Q2 zone) and the rest of the system is modeled with molecular mechanics (MM). As the substrate moves, the atoms in the system are reclassified into the appropriate zone on the fly during molecular dynamics simulations. Buffer zones between adjacent layers allow a smooth extrapolation of energies and forces. This project combines method development, software implementation (GAMESS and QMMM) and application simulations.
Prediction and analysis of sigma-hole interactions in molecular crystals.
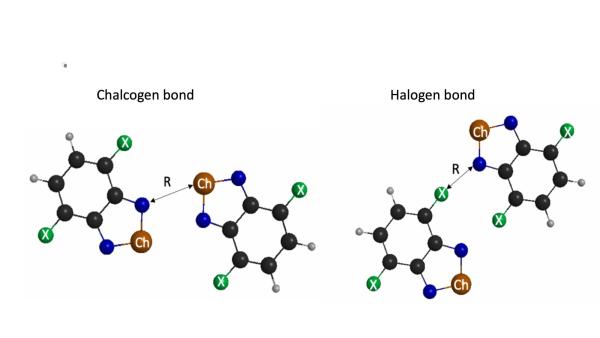 Predicting and understanding intermolecular forces is essential for the design of tunable nanostructures or even drug delivery systems. In collaboration with Dr. Christer Aakeröy at Kansas State University, our group uses ab initio methods and energy decomposition schemes to predict and analyze sigma-hole interactions in molecules and crystals. Examples of sigma-hole interactions include:
Predicting and understanding intermolecular forces is essential for the design of tunable nanostructures or even drug delivery systems. In collaboration with Dr. Christer Aakeröy at Kansas State University, our group uses ab initio methods and energy decomposition schemes to predict and analyze sigma-hole interactions in molecules and crystals. Examples of sigma-hole interactions include:
- Halogen bonding: the interaction between a chemically bonded halogen atom (X=fluorine, chlorine, bromine,iodine) and a nucleophile such as the nitrogen of ammonia.
- Chalcogen bonding: the interaction between a chemically bonded chalcogen atom (Ch=oxygen, sulfur, selenium, tellurium) and a nucleophile.
Sigma-hole interactions have opened new venues in molecular design as they can be as strong and directional as hydrogen bonds. They are also highly tunable. Our group is interested in understanding the electronic interactions responsible for the formation of these bonds and determine the importance of covalent forces.
Prediction of host-guest and protein-ligand binding affinities.
Synthesizing and testing chemical drugs is a long and expensive process, especially when hundreds of compounds need to be tested. Therefore, pharmaceutical companies use computational methods to screen potential drug candidates, therefore significantly narrowing down the number of compounds to be synthesized. We are interested in developing new computational methods to predict the binding free energy of protein-ligand complexes.These new methods combine conformational search algorithms (VM2) with classical molecular mechanics and accurate quantum mechanical methods. This work is in collaboration with Vera Chem LLC and the Gordon group at Iowa State University.
Reduction of carbon dioxide on thiolate protected gold nanoclusters.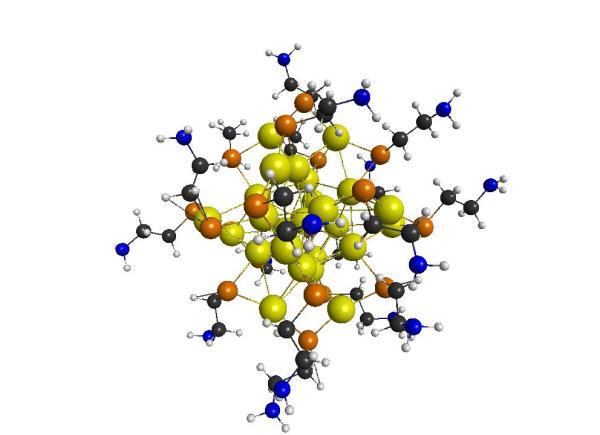
Thiolate-protected nanoclusters have shown great potential in catalysis, including the reduction of carbon dioxide (a green house gas) into a syngas mixture. We are interested in identifying how the ligand shell of the nanocluster and the solvent environment affect the reduction pathway of carbon dioxide. In addition, we are also interested in developing more efficient computational methods to model the electronic properties of these systems and their interactions with solvent molecules.